Currently Available SRA IDs
We currently have 249,034 runs from the sequence read archive preprocessed and ready for searching. You can retrieve a list of all of the SRA IDs. These IDs were identified by PARTIE as being most likely metagenomic runs.
We have also provided a set of IDs for the TARA Oceans Project and the Human Microbiome Project. These sets are smaller, and thus will process faster.
As you will recall, a project (SRP) has one or more samples; a sample (SRS) has one or more experiments (SRX); and an experiment has one or more runs (SRR), and in searchSRA we use the SRR IDs as the primary key.
We have combined all the SRR IDs that we have available in searchSRA and provide them as a tab-separated text file that connects the runs with 18,599 projects (SRP identifiers) and includes the title and abstract. You should be able to open this file in a Excel, LibreOffice, or a similar spreadsheet program, as well as Python or R.
Of course, you can always find more about the SRA Metadata and Hidden SRA Metadata from our blogs.
Tools to analyze the data
We have created some tools to help you analyze the data that you generate at SearchSRA.org.
There are detailed instructions at the Git Repository, but essentially we recommend using the filter_reads.sh script that will do everything for you.
Check the Git Repository, and be sure to cite our work!
Training and Documentation
Search SRA Gateway User Manual
Gateway Access
- Create a gateway account at https://www.searchsra.org/. Use 'Create account'. Provide your details and you will receive an email for account verification.
- Click the link in the email received and confirm your email. Hint: The email sometimes tends to land on 'Junk email' or 'Trash'.
- When the gateway administrator grants access, you would receive a notification email.
- Log in to the gateway, You are ready to Search through the SRA!
Getting Started
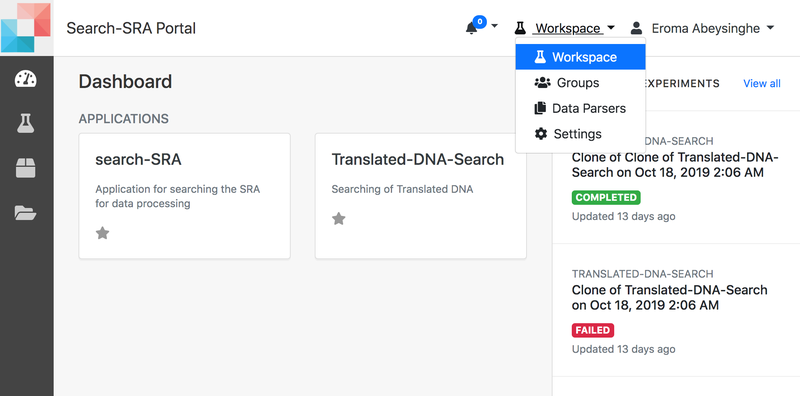
- When logged in you would be on your home page, Workspace.
- You would see your application placeholders.
- Choose the one you want to use and click to navigate to the 'Create Experiment' page.
- To your right, you would see all your recent experiments.
- On the top-right menu, you will have
- Workspace (you are there now)
- Groups
- Settings
- The left vertical menu is the child menu of what you select from the above, right-hand drop-down menu.
- Workspace has
- Dashboard
- Experiments
- Projects
- Storage
- What is an Experiment? - Experiment is the gateway record you create in order to launch a job in a remote cluster/supercomputer/HPC.
Save & Launch an Experiment
- To create a new Experiment from the Dashboard select your application.
- In Create Experiment default values are provided for the name.
- Experiment Description (Optional)
- Project (LOV) - Last used will be selected for you
- Application (LOV)
- Application configurations will have default set values with option to change.
- Application Inputs (Mandatory)
- Save & Launch
- When providing Application inputs;
- Fasta-Reference-File (Mandatory)
- Select existing Search IDs File OR provide your own.
- After providing the files you could 'Save' the experiment for later submission. If saved you could edit the experiment prior to launching.
- Once launched, you could cancel the experiment, which will cancel the job running in the remote cluster.
Other Gateway Features
- Users can share their experiments with other gateway users or user groups. When sharing, you can decide on the level of access to grant.
- Read Access
- Write Access
- To create new experiments similar to an existing one, you can clone the experiment from Experiment Summary page.
- Users can create their own user groups to share research experiments, data files.
- Group Resource Profile of the user enables to user to use their own research allocation/logins for computations.
What do I do if I only find a few or no results?
If you are performing DNA searches, it maybe that the DNA sequence is too diverged for this to match using bowtie. There are two suggestions:
- Try using the protein sequence and running a protein search on searchsra.org.
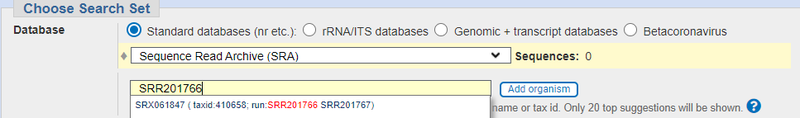
- For a single SRR, you can run blast at the NCBI BLASTN website. Choose Sequence Read Archive from the database pull down, and then put your SRR ID in. It will automagically pull up the SRX number for you that blast needs.
When I tried this with an example sequence, I got three hits with not very good E-values:
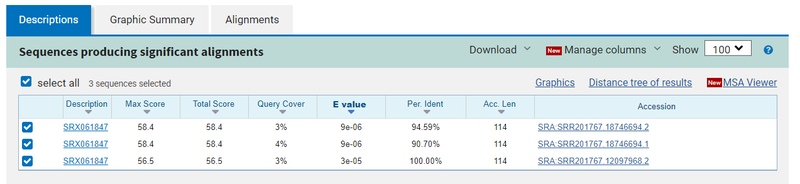
Recall that in searchSRA one of our heuristics is to only search 100,000 sequences. That data set has 10<sup>10</sup> sequences, so if only 3 sequences in 10<sup>10</sup> match, we would not report that in searchSRA any way.
- You can also use the TBLASTN suite at NCBI to search a translated SRA run against a protein. I used the EXPASY Tool to translate my DNA sequence to protein, and compared it to the same run using TBLASTN and got more than the 100 maximum number of hits with better E values:
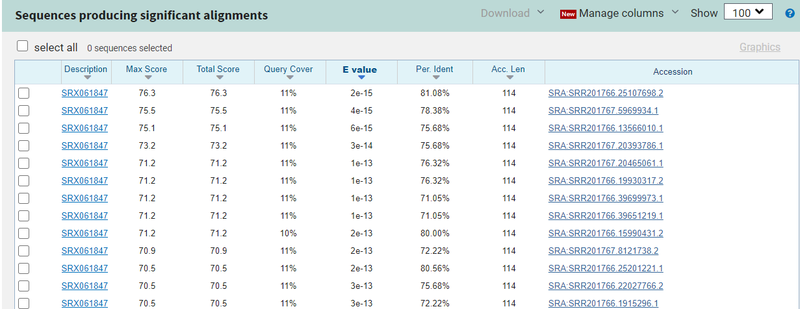
This would suggest solution #1 (using protein searches) would be a better option for searching the entire SRA (which, alas, you can not do at NCBI!)